歐盟醫療器材 MDR / IVDR 法規正式實施 歐盟醫療器材 MDR / IVDR 法規正式實施
醫療器材業者需特別注意,自 2021 年 5 月 26 日起歐盟醫療器材法規改革,所有進入歐盟市場醫療器材,均須符合醫療器材法規(MDR)所設定的標準,否則將無法進入市場,體外診斷醫療器材法規(IVDR) 自 2022 年 5 月 26 日起適用。這項改革大幅墊高醫療器材製造廠商進入歐盟市場門檻,新法規要求審查標準與產品技術發展同步提升,並加強產品安全、性能評估、臨床驗證及上市後監管。此外,技術審查變得更加嚴格,供應鏈的可追溯性要求也大幅提升,以確保產品在市場上的安全性與可靠性。
我們的歐洲夥伴已獲當地衛生主管機關 HPRA 核可,具備授權代表與進口商資格,能夠提供 MDR / IVDR 歐盟授權代表及法定進口商服務,公司自成立之初便專注於法規與醫療器材進口服務,擁有多年輔導實務經驗,專門協助台灣廠商合法將醫療器材投放至歐盟市場。此外,可以中文溝通,確保交流順暢,降低法規適應門檻,讓您的產品更快速符合歐盟要求。

歐盟關於醫療器材與體外診斷試劑的新法規 MDR / IVDR 概述
歐盟於2017年頒布了醫療器材和體外診斷的新法規,以適應過去 20 年該產業的發展。新的醫療器材法規 (EU) 2017/745 (MDR) 和體外診斷醫療器材法規 (EU) 2017/746 (IVDR) 使歐盟立法與現今醫學及科技的進步保持一致。與指令不同,法規可直接適用,毋需轉化為國家法律。因此,MDR 和 IVDR 能降低歐盟市場因為國家法令詮釋差異造成的風險。
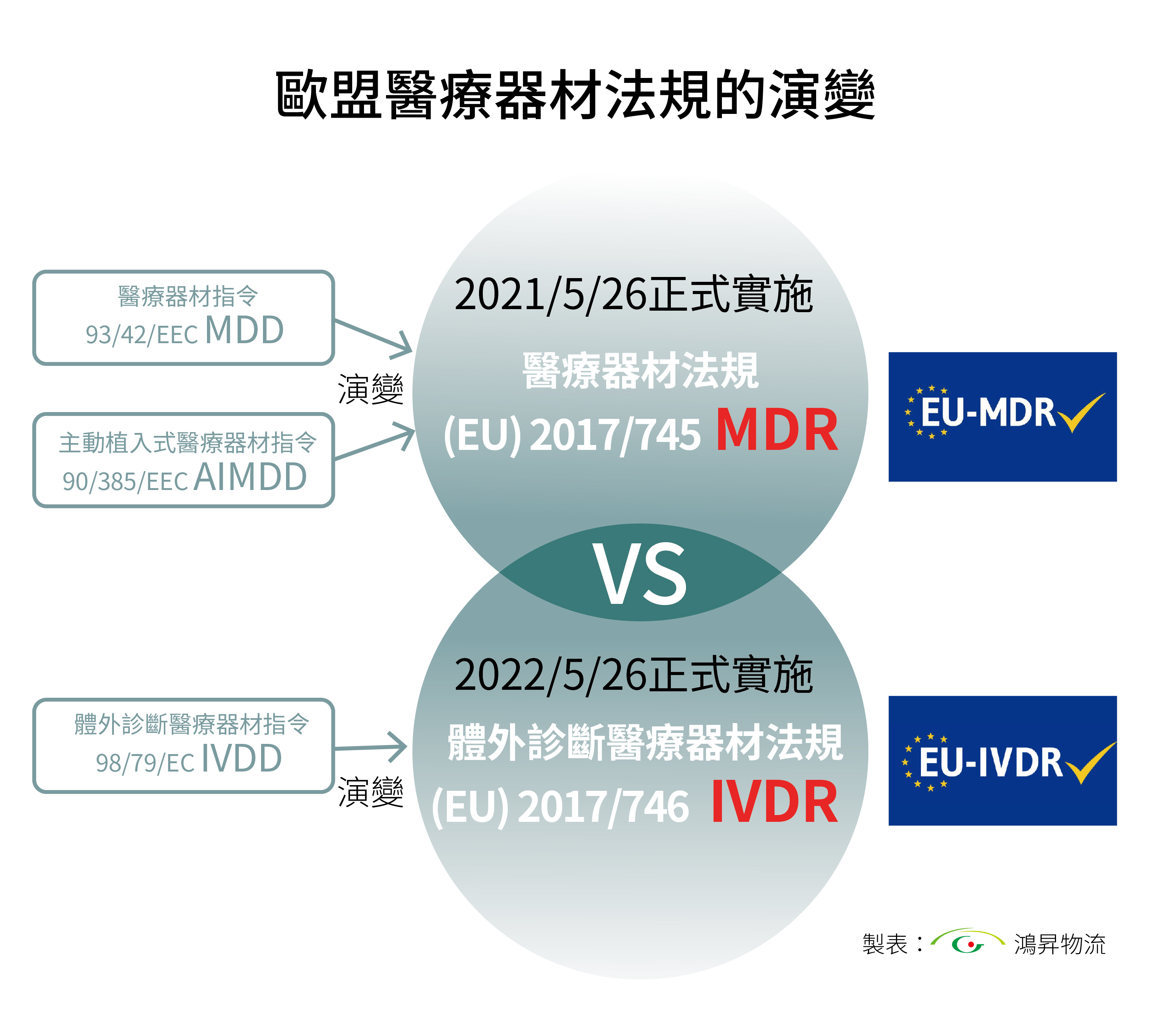
這兩項關於醫療器材和體外診斷醫療器材的新法規已於2017年5月生效。MDR將取代現有的醫療器材指令 93/42/EEC(MDD)和主動植入式醫療器材指令 90/385/EEC(AIMDD);IVDR將取代現有的體外診斷醫療器材指令(98/79/EC)(IVDD)。 這兩個法規皆規定了過渡期,在此期間符合先前指令的設備仍可投放到歐盟市場。時至今日,隨著轉換期接近尾聲,歐盟市場已逐漸要求醫療產品須帶有符合 MDR / IVDR 規定的CE標誌。
角色與責任
— 授權代表的角色與責任
關於授權代表的一般義務羅列於 MDR / IVDR 第11條。授權代表是在歐盟內設立的任何自然人或法人,接受非歐盟製造商的書面委託,代表製造商執行與其法規責任相關的特定任務。 MDR / IVDR 第11條法規規定製造商須委託授權代表執行的任務,應以書面授權委託書來規範。
| 授權代表的基礎責任包括 |
| 1. |
確認製造商已經編製了歐盟符合性聲明和技術文件,並在適用的情況下,製造商已執行相應的符合性評估程序( MDR / IVDR 第11條第3項(a))。 |
| 2. |
授權代表還必須保留所有文件的副本並根據要求向當局提供,這包括技術文件、符合性聲明和EC證書,包含其修訂與補充文件( MDR / IVDR 第11條第3項(b))。 |
| 3. |
授權代表必須核實製造商是否已在EUDAMED註冊所要求的信息( MDR / IVDR 第11條第3項(c))。 |
| 4. |
授權代表需與當局合作處理預防與矯正措施,並立即向製造商反映投訴以及當局對樣品的要求。若製造商未履行其在法規下的責任,且製造商不在歐盟境內,授權代表需與製造商一同對有缺陷的設備負責( MDR / IVDR 第11條第5項)。 |
| 5. |
若製造商違反MDR法規義務,授權代表應終止授權委託(MDR / IVDR 第11條第3項(h))。在此情況下,授權代表應立即通知其所屬成員國,說明終止授權委託的原因。若產品經公告機構進行合格性評估通過,則需一併通知該公告機構。 |
法規還規定不得委託給授權代表的活動( MDR / IVDR 第11條第4項)。例如,涉及設備設計、品質管理系統或技術文件編製的要求,這些是製造商的專屬責任。此外,授權代表應由有經驗的法規人員 PRRC (Person Responsible for Regulatory Compliance)負責所有合規事務( MDR / IVDR 第15條第6項)。最後,更換授權代表需要有一個適當的協議來界定製造商與離任及新授權代表之間的安排( MDR / IVDR 第12條)。
— 進口商的角色與責任
關於進口商的一般義務羅列於 MDR / IVDR 第13條。進口商定義為在歐盟設立的任何自然人或法人,將來自非歐盟製造商的設備投放至歐盟市場。
| 進口商的的責任包括 |
| 1. |
確保其投放市場的設備標有CE標誌,附有所需的信息並依照法規標註,並在適用的情況下,設備已有單一識別 (UDI) 碼。 |
| 2. |
進口商應驗證設備是否已在 EUDAMED 註冊。 |
| 3. |
如果進口商認為某設備不符合法規,該設備不得投放市場,並且進口商應通知製造商和授權代表。若進口商懷疑設備遭偽造或存在嚴重健康風險,應立即通知主管機關。 |
| 4. |
進口商應確保在其負責的儲存與運輸條件下不會危及合規性。進口商應在設備或其包裝上,或設備隨附的文件中標示其名稱、註冊商號或註冊商標、註冊商業地址及聯繫方式。 |
| 5. |
進口商在收到投訴時應負責通知製造商及其授權代表。 |
| 6. |
進口商應對投訴、不合格設備、召回與撤回等市場活動進行紀錄。此外,若懷疑設備遭偽造或存在嚴重健康風險時,應將不合規事項呈報主管機關。 |
| 7. |
進口商還需要配合主管機關,提供樣品或使其能夠接觸設備。 |
提供的服務
我們的歐洲夥伴獲得當地衛生主管機關 HPRA 核可具備授權代表資格與進口商資格,能滿足台灣醫療器材製造商的合規需求。我們的服務旨在確保所有品質、法規和技術文件符合 MDR 和 IVDR 標準,從而促進順利進入歐盟市場。我們提供專業服務,確保貴公司產品合規且快速地進入歐洲市場:


參考資料
- MDR 法規本文 https://eur-lex.europa.eu/eli/reg/2017/745/oj/eng
- IVDR 法規本文 https://eur-lex.europa.eu/eli/reg/2017/746/oj/eng
- MDCG Guidance 文件清單 https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en

線上諮詢
若您需要醫療器材提供之歐體代表與法定進口商服務,歡迎來信諮詢!
|